
作者信息
第一作者:Jordi Ballesteros-Soberanas
通訊作者:Donatella Armentano, Emilio Pardo, Antonio Leyva-Pérez
通訊單位:瓦倫西亞大學、加的斯大學等
成果速覽
本研究展示了一種定義明確的Pd1-Au1二聚體,錨定在金屬-有機框架(MOF)的壁上,能夠在模擬工業前端反應條件下,選擇性地將乙炔半加氫爲乙烯,轉化率≥99.99%(乙炔≤1 ppm)且選擇性>90%。 反應在35°C甚至更低的溫度下進行,操作窗口>100°C,重量小時空間速度(WHSV)爲66,000 ml g^-1 cat h^-1,符合工業規格。 實驗和計算機制研究顯示,兩個原子之間的協同作用以及與支撐材料的相互作用,實現了乙炔的無障礙半加氫。
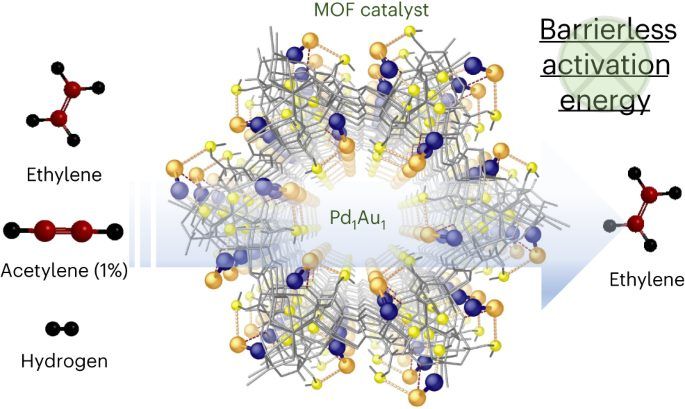

圖1:展示了Pd1-Au1二聚體在MOF中形成的過程,以及通過X射線晶體結構測定得到的Pd1Au1@1的結構細節。

圖2:展示了使用Pd1Au1@1作爲催化劑進行乙炔半加氫反應的催化實驗結果。
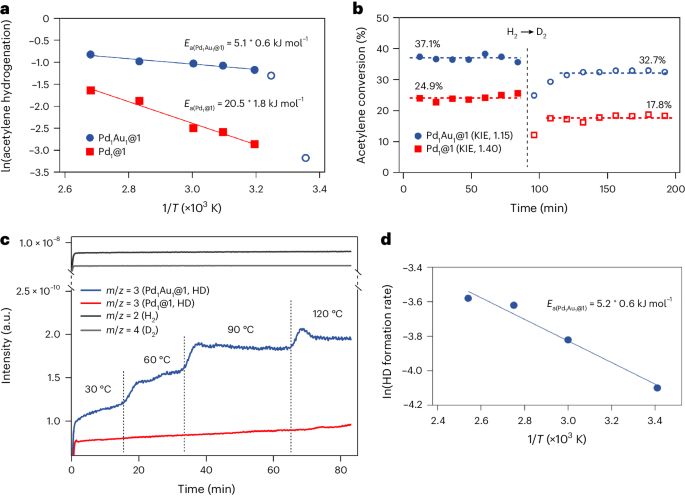
圖3:提供了Pd1Au1@1和Pd1@1的實驗機理證據。

圖4:理論計算提供了乙炔加氫反應的最可能的中間體和提出的機制。
亮點介紹
1. Pd1-Au1二聚體催化劑在極低的活化能(約1 kcal/mol)下實現了乙炔的高效半加氫,轉化率超過99.99%,且乙烯選擇性高于90%。
2. 催化劑在35°C至150°C的溫度範圍內均表現出優異的催化活性,且在工業相關操作條件下具有較長的穩定性。
3. 通過結合實驗和計算研究,揭示了Pd1-Au1二聚體與MOF之間協同作用的機制,爲工業反應提供了新的催化策略。
4. 該研究提供了一種新型的高分散、高金屬負載的催化劑設計方法,有助于推動石化工業中關鍵反應的能效提升。
計算模擬
在本論文中,作者進行了詳細的計算化學研究,以支持和解釋實驗觀察到的催化行爲。 通過使用密度泛函理論(DFT)計算,研究人員對Pd1-Au1二聚體在MOF中的催化活性位點進行了優化,並探究了其電子結構。這些計算幫助確定了最穩定的Pd1-Au1二聚體結構,並預測了其與MOF中硫醚基團的相互作用。
文獻信息
標題:A MOF-supported Pd1–Au1 dimer catalyses the semihydrogenation reaction of acetylene in ethylene with a nearly barrierless activation energy
期刊:Nature Catalysis
DOI:10.1038/s41929-024-01130-7
