
編者按:細胞凋亡是生物體正常發育和維持穩態的重要過程。生理狀態下,它參與了發育過程中非必要結構的退化以及多余細胞的清除;病理狀態下,細胞凋亡調控的異常促進了腫瘤、神經系統以及自身免疫性疾病的發生,並與放化療抵抗相關,因此針對腫瘤細胞凋亡逃逸的研究已成爲當下的熱門。
本期「專家組稿」由複旦大學附屬眼耳鼻喉科醫院王孝深教授擔任執行主編,與複旦大學附屬腫瘤醫院許婷婷教授分享IAP抑制劑研究進展,爲醫者和患者提供更多參考。
專家介紹

王孝深
複旦大學附屬眼耳鼻喉科醫院放療科主任醫師、博士研究生導師
上海市抗癌協會鼻咽癌專業委員會副主任委員
腫瘤影像與康複治療專家委員會副主任委員
中國抗癌協會鼻咽癌專業委員會常務委員
中國醫師協會頭頸腫瘤專業委員會常務委員
中國臨床腫瘤學會鼻咽癌專家委員會常務委員
中國醫藥教育協會頭頸腫瘤專委會常務委員
中國抗癌協會鼻咽癌整合康複專委會常務委員
上海市抗癌協會理事
中國臨床腫瘤學會頭頸腫瘤專家委員會委員
中國抗癌協會頭頸腫瘤專業委員會委員

許婷婷
複旦大學附屬腫瘤醫院
副主任醫師、博士
中國抗癌協會鼻咽癌專委會常委
CSCO頭頸腫瘤專委會委員兼秘書
中華醫學會鼻咽癌學組委員
國家癌症中心鼻咽癌質控專委會委員兼秘書
中國抗癌協會神經腫瘤專委會青年委員
上海市抗癌協會第二屆青年理事會理事
《中國癌症志》第五屆編委會青年編委
上海市醫學會腫瘤放射治療委員會青委兼秘書
題目:IAP抑制劑研究進展
◾ 王孝深 許婷婷 複旦大學附屬眼耳鼻喉科醫院/複旦大學附屬腫瘤醫院
細胞凋亡的途徑可以分爲caspase依賴性和非依賴性,其中caspase依賴性包括由受體配體結合起始的外源性途徑(起始caspase 2,8,9,10可自我剪切;效應caspase 3,6,7需由其他caspase來剪切),以及促使線粒體釋放細胞色素C等細胞因子,與Apaf-1結合後激活起始caspase-9再切割效應caspase-3的內源性途徑;caspase非依賴性途徑主要通過線粒體釋放AIF和Endo G進入細胞核,切割核DNA誘導凋亡。caspase是一種蛋白水解酶,必須與相應的底物相結合以後才能發揮促凋亡的作用,它主要的效應包括釋放DNA內切酶,失活DNA損傷修複酶PARP以及促進凋亡小體的形成。
一、凋亡抑制蛋白(Inhibitor of Apoptosis Protein, IAP)抑制劑放療增敏的機制
凋亡抑制蛋白(Inhibitor of Apoptosis Protein, IAP)家族是一組凋亡負調控因子,包括8個家族成員,它的特征是在其肽鏈的n端存在至少一個杆狀病毒IAP重複序列 (BIR)結構域。目前研究比較透徹的是XIAP、cIAP1和cIAP2,其中XIAP可以結合caspase-3、7、9直接發揮阻斷作用,具有最顯著的抗凋亡活性。而cIAP1、cIAP2則是通過抑制促凋亡複合物的形成,實現對凋亡通路的調控。三個IAP都具有三個BIR結構域,C端還具有RING結構域。RING具有泛素蛋白連接酶(E3)活性,通過泛素化凋亡調控因子,關閉細胞凋亡途徑。
IAP與caspase CIAP-1和c-IAP2還具有caspase-recruitment domain (CARD)結構域,參與E3活性的調控。作爲caspase的抑制劑,IAP一方面可以通過物理阻斷掩蓋caspase的活性位點,阻止底物進入,還可以靶向caspase蛋白酶體降解。其中XIAP是嚴格意義上唯一的直接caspase抑制劑,它的BIR3選擇性地靶向caspase-9,而BIR2結構域及其連接子可以同時結合caspase-3和7,在凋亡抑制通路中發揮核心作用。cIAP則是通過RING結構域的E3連接酶活性,使caspases-3和-7的蛋白酶體降解。另外還參與了NF-kB通路的調控。
IAP與NF-κB信號通路 NF-κB家族包括五個成員,它們可以以二聚體形式被活化後促進下遊的轉錄因子激活。經典NF-κB通路是在胞外刺激(射線、藥物,配體受體結合,TNFα)的作用下,使IKK(IkB激酶複合物)激活,IkB蛋白降解,釋放NF-κB二聚體,轉移至細胞核,促進目的基因轉錄。而非經典 NF-κB 通路則是通過激活NIK,磷酸化IKKα使p100轉化爲p52,産生激活的NF-κB複合物,轉移到細胞核並誘導下遊基因表達。IAPs是NF-κB信號通路的關鍵調節因子。cIAP通過對RIPK1的泛素化降解,限制了促凋亡複合物II的形成,促進複合物I形成,激活促存活的經典NF-κB。另外,cIAPs還能使NIK發生泛素化降解,阻斷非經典通路的激活,抑制炎症細胞因子如TNFα的釋放。反過來,IAPs的缺失導致促凋亡複合物II的形成並激活caspase8介導的凋亡,同時通過NIK的積累,激活非典型通路,促進炎症因子的釋放。IAP抑制劑能通過競爭性結合IAP的BIR結構域,來解除IAP對caspase的抑制作用,成爲目前最被看好的靶向凋亡的藥物之一。
IAP與Smac蛋白 人體存在非常微妙的平衡,在體內發現了一種與IAP相互作用蛋白,稱爲第二線粒體來源的caspase激活因子Smac(Second mitochondria-derived activator of caspases),當細胞受到凋亡刺激時,線粒體釋放Smac到細胞質,後者與IAPs結合,使其喪失抑制caspase活性的作用,從而促進細胞凋亡。Smac的作用除了中和IAP蛋白外,可以誘導IAP的E3鏈接酶活性,從而導致其自泛素化而降解。內源性SMAC蛋白與XIAP的BIR3的親和力高于BIR2,因此對caspase9的解除作用更強。
但Smac作爲一種較大的蛋白質,在臨床使用時可能會産生嚴重的脫靶效應。研究發現,Smac N端的四肽序列AVPI是主要的與BIR結構域結合的部位,AVPI片段也是保留Smac抑制活性的最小片段,因此采用化學合成序列相似的小分子化合物Smac類似物(IAP抑制劑),也能達到抑制IAPs的目的。IAP抑制劑可同時阻斷XIAP和cIAP,通過促凋亡和抑制NF-κB通路兩方面發揮抗腫瘤作用。放療主要通過誘導凋亡(間接作用)和抑制有絲分裂(直接作用)發揮抗腫瘤作用,誘導凋亡在放療抗腫瘤機制中占重要地位,而化療可能更多依靠抑制有絲分裂或其他途徑(如抗代謝)。電離輻射可以通過降解IkB激活NF-κB經典通路,並誘導TNF-α上調,同時去除TRAF-2或cIAP-1可以激活由TNF-a啓動的死亡受體通路從而誘導凋亡。研究顯示,Smac類似物SM-164可以增強輻射誘導的NF-κB,導致TNF-a轉錄和分泌增加,促使caspase-8激活。聯合IAP抑制劑可以降低放射治療誘導的細胞死亡的阈值實現放療增敏。
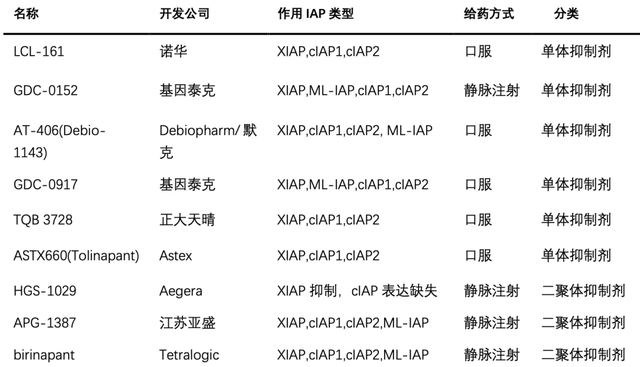
Smac類似物的臨床研究現狀 目前已經進入臨床研究階段的Smac類似物主要有兩種類型,一種是單體抑制劑,其與IAP只有1個結合位點,能與cIAP-1/2 和XIAP的BIR3結構域結合,從而Caspase-9得以釋放;另一種是二聚體抑制劑,有2個結合位點,能與cIAP-1/2的BIR3域以及與XIAP的BIR2和BIR3域同時結合,從而同時釋放Caspase-3、7、9。實驗證明二聚體具有更高的親和力,但是單體可以口服,具有更高的生物利用度,而二聚體需要通過靜脈注射給藥。
表1.已進入臨床研究階段的IAP抑制劑
二、IAP抑制劑在實體瘤中放療增敏的研究現狀
IAP抑制劑在實體瘤中的探索方向主要是聯合放療用于頭頸部鱗癌或鼻咽癌,在其他瘤腫中的研究包括和PD-1單抗或化療聯合。
表2.IAP抑制劑治療實體瘤在研項目

目前在局部晚期頭頸鱗癌中已經布局的IAP抑制劑與放療聯合的項目有4個,涉及的藥物有2個:包括默克的AT-406(Debio-1143)和Astex制藥公司的ASTX660。
作用機制 Debio-1143可阻斷XIAP和cIAP1/2,從而解除凋亡抑制,抑制XIAP可直接釋放內源性通路中下遊的caspase活性,而抑制cIAP1/2可通過外源性通路促進TNF介導的促凋亡信號,腫瘤微環境中的TNF-α可以與之形成正反饋並放大凋亡信號,但外源性TNF-a的全身給藥毒性非常大,使其無法得到推廣。
聯合化療 Debio 1143的臨床前研究顯示其單藥的抗增殖能力有限,而聯合卡鉑和順鉑能誘導頭頸癌患者腫瘤樣本産生caspase-3依賴性凋亡,認爲Debio 1143聯合化療可能是一種潛在治療方法。體外實驗證明,放射治療能誘導TNF-α的産生,反過來,TNF-α又增強輻射介導的殺傷。對藥物和放療的最佳聯合模式的探索也證實Debio 1143與RT同步使用,並進行10天的維持,可以達到最佳療效,因此提示臨床應考慮與放療同步聯合輔助治療。研究表明,Debio 1143聯合RT在誘導腫瘤細胞凋亡的同時還伴隨著TNF-a mRNA水平的顯著升高,采用抗TNF-a抗體來中和TNF-a可以減弱聯合治療的抗增殖能力,強烈提示TNF-a在Debio 1143誘導的放射增敏中的作用。
臨床研究 Xevinapant在局部晚期頭頸鱗癌中與放化療聯合的2期RCT研究已經有長期陽性結果報道,在基于標准劑量順鉑CRT的基礎上增加3周期的口服Debio 1143來增敏放療,可使死亡風險下降53%。5年OS達到53%,接近標准CRT的2倍(HR 0.47, 95% CI 0.27-0.84, P=0.101)。在其III期的拓展隊列研究中,方案在放化療結束後又增加了三個周期單藥的維持治療,全球預計共入組700例,目前研究仍然在進行。另外一項針對具有高危因素的、鉑類不能耐受患者的術後輔助放療同步采用Xevinapant增敏的III期研究也在積極地入組中。
三、IAP抑制劑與免疫檢查點抑制劑(ICIs)聯合
早期的研究已經證明IAP和PD-L1能共同保護腫瘤細胞免受源自CD8+T細胞的促凋亡作用,兩者的共同抑制作用顯著提高了腫瘤細胞對細胞毒作用的敏感性阈值,是重要的耐藥機制之一。由于IAP抑制劑對于IAP的阻斷作用還受到TNF-α水平的影響,其會以TNF依賴的方式發揮作用,通過IAP抑制劑來調控cIAP蛋白一方面可以促進經典通路下caspase8依賴的凋亡,另一方面也可以通過泛素化降解消耗cIAP蛋白,使得非經典通路的核心蛋白NIK更穩定,進而激活非典型通路,進一步誘導TNF-α表達,參與自分泌/旁分泌,形成正反饋,並促進細胞進入死亡循環。有研究觀察到當三陰性乳腺癌的腫瘤微環境中缺乏TNF-α時,腫瘤細胞將表現爲對IAP抑制劑治療耐藥。基于這一理論,阻斷PD-1/PD-L1可以增加效應細胞分泌TNFα的水平,因此ICIs可以和IAP抑制劑産生協同作用。
Cell雜志上發表的一項研究同樣顯示,ICIs、CAR-T細胞或溶瘤病毒可以誘導死亡的腫瘤細胞釋放TNF,聯合IAP抑制劑可以激活TNF自分泌信號通路,介導對于無抗原表達的腫瘤細胞的旁觀者殺傷。另外,IAP抑制劑還具有調節先天和適應性免疫的作用,通過抑制cIAP1和cIAP2,激活非經典NF-κB通路,可以促進B細胞存活,激活樹突狀細胞,並向T細胞傳遞廣泛的共刺激信號。也能使巨噬細胞從免疫抑制型M2型向促炎型M1分化,促進TNF-α分泌。
目前已有多項IAP抑制劑與ICIs聯合治療的臨床研究,當然大部分還是在I期階段(表3),在頭頸癌領域的探索包括了在鼻咽癌中的江蘇亞盛的APG-1387與特瑞普利單抗的聯合以及局晚或轉移SCCHN中的Birinapant與帕博利珠單抗的聯合,但後者因爲在結直腸癌隊列已經達到無效終點而關閉了研究。
表3.IAP抑制劑與ICIs聯合治療研究

Clinical Cancer Research雜志今年發表的一項後線藥物治療晚期結直腸癌(CRC)和胰腺癌(PDAC)的研究,是首個有完整臨床數據的報道IAP抑制劑和ICI聯合用藥研究,顯示入組的40個患者中只有1個達PR,4個SD,且都是胰腺癌患者,有效率低。觀察到cIAP-1表達水平在用藥後顯著降低,表明低于預期的療效並非由于藥物暴露不足導致。在大多數CRC患者中檢測到caspase-3的激活,但在PDAC中卻是相反的現象,提示IAP抑制劑的抗腫瘤作用在PDAC中存在的潛在代償機制。類似的聯合用藥,在一項Xevinapant+O藥治療多瘤種的1b/2期研究中,顯示ORR只有2.9%,另一項入組非小細胞肺癌的研究中采用Xevinapant+阿維魯單抗,去年AACR會議上報道了ORR也只有10.5%,與曆史數據對照,沒有超過阿維魯單抗的單藥有效率。
盡管IAP抑制劑與ICIs聯合具有良好的理論基礎和臨床前數據,但現有的臨床試驗結果仍然令人失望,初步報道均沒有顯示出兩者聯合能進一步提高治療有效率。因此還需要進一步探索合適的聯合時機、用藥劑量以及是否需要其他療法的參與。

版權聲明
本文版權歸醫悅彙所有。歡迎轉發分享,其他任何媒體如需轉載或引用本網版權所有內容,須獲得授權,且在醒目位置處注明“轉自:醫悅彙”。

